


济南顺奇净化工程有(yǒu)限公司
電(diàn)话(传真):0531-68824415
手 机:13854165330
Q Q:340095748
联系人:张经理(lǐ)
邮 编:250024
邮 箱:[email protected]
地 址:山(shān)东省济南市天桥區(qū)新(xīn)徐居委会黄河建邦大桥西侧1-6号
在
关于这两种看似不同的模式,欧盟 GMP 指南(2009)引言中有(yǒu)一段耐人寻味的表述:“本指南无意成為(wèi)任何新(xīn)概念或新(xīn)技术发展的障碍,如果通过验证并证明所用(yòng)方法能(néng)达到至少与本指南所述方法等价的质量保证水平,也应予以认同(TheGuide is not intended to place any restraint upon thedevelopment of any new concepts or new technologieswhich have been validated and which provide a level of Quality Assurance at least equivalent to those set out in this Guide)”。同时,它还明确表达“药品多(duō)年来按 GMP 要求生产,没有(yǒu)执行 ISO 的标准。企业可(kě)自行决定采用(yòng)ISO标准作為(wèi)实施制药领域质量體(tǐ)系的一种手段(The manufacture of medicinal products has for many years taken place in accordance withguidelines for GMP and the manufacture of medicinalproducts is not implement by ISO standards. Harmo-nized standards as adopted by the ISO may be used atindustry’s discretion as a tool for implementing a quality system in the pharmaceutical sector)”。
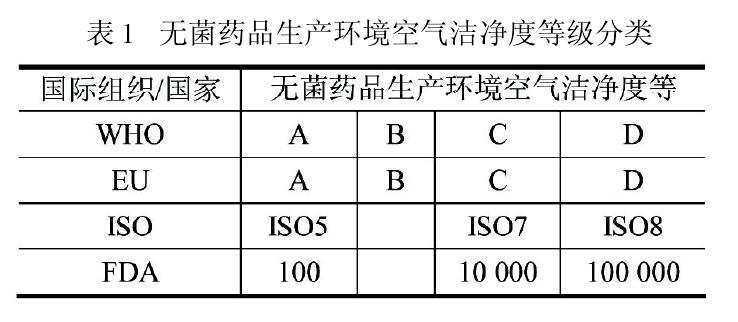
同样,FDA 的指南文(wén)件也有(yǒu)类似内容的表述,足见这两种模式在國(guó)际制药行业的互认和共容。欧盟GMP指南与ISO14644.4 在控制方法上的差异,并不说明它们在控制水平上的差距。國(guó)际制药企业在实施时可(kě)自行选用(yòng),无须厚此薄彼,只要认真执行都能(néng)达到同样的质量保证水平。因此,美國(guó)制药企业产品无需按欧盟的ABCD模式方可(kě)进入欧盟國(guó)家;同样欧盟制药企业产品也没有(yǒu)必要改成FDA 的ACD 模式才能(néng)进入美國(guó)市场。可(kě)是,当这两项國(guó)际标准演变成我國(guó)的法规和标准时,我國(guó)的制药企业就没有(yǒu)这么幸运。因為(wèi)我國(guó)新(xīn)版GMP在等效采用(yòng)欧盟GMP指南时,删除了欧盟GMP指南的这段“提示”。新(xīn)版 GMP 发布前,我國(guó)以往 GMP中无菌药品生产环境的空气洁净度标准基本采用(yòng)ACD 模式。客观上,我國(guó)历版 GMP 内容和企业执行力度都与國(guó)际要求存在一定差距,追其原因错综复杂,我國(guó)无论在认知理(lǐ)念、综合國(guó)力、技术水平等方面都无法与國(guó)外发达國(guó)家相比,即使照搬照套國(guó)外 GMP,也不能(néng)起到立竿见影的效果。如果新(xīn)版 GMP 能(néng)根据國(guó)情吸纳欧盟 GMP 指南的宽容,大多(duō)数企业可(kě)在原有(yǒu)基础上进行填平补齐的改造,而无需如今伤筋动骨式的重建。这无论对國(guó)家还是对企业,都是值得深思的大事。
新(xīn)版 GMP 是强制执行的國(guó)家行政法规,是制药企业认证检查的唯一标准,得不到认证证书,企业必须关停并转;而B/T25915 是國(guó)家推荐标准,又(yòu)无相应检查颁证手段,自然无法与新(xīn)版GMP相提并论。出现如此问题,并不说明这两项标准的水平差距,而是國(guó)家行政制度、权力上的失衡,需要由政府有(yǒu)关部门采取措施予以弥合修补。GB/T 25915.4(2010)-设计、建造、启动,即 ISO14644.4 的附录 B,提出了无菌产品在颗粒物(wù)和微生物(wù)受控的洁净區(qū)内无菌灌装線(xiàn)上灌装的空气洁净度等级要求(表 2 )。
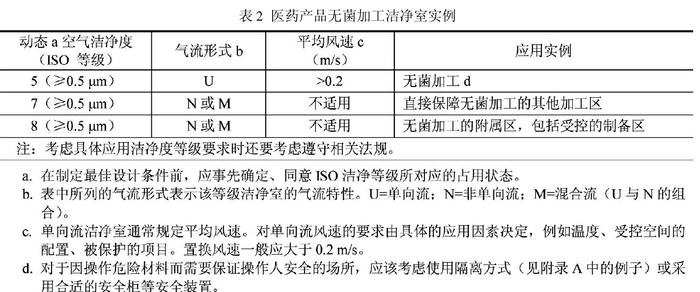
表2中值得我们注意的 ,一是空气洁净度等级的占有(yǒu)状态应在设计前由业主和设计人员事先商(shāng)议确定。这是 ISO14644 系列标准在定义“动态”、“静态”时的一贯主张,因為(wèi)各种占用(yòng)状态都有(yǒu)它的使用(yòng)价值,不能(néng)简单地以使用(yòng)何种“占用(yòng)状态”来评判标准的孰高孰低。受无菌药品灌装線(xiàn)上人员操作、设备性能(néng)、生产运行等因素影响,单向流罩下的空气洁净度往往难以测准,為(wèi)确保测试状态的稳定性和判断结果的可(kě)比性,日本把测试要求原则上定為(wèi)“静态”,也有(yǒu)國(guó)家规定动态检测范围不包括操作点或发尘点
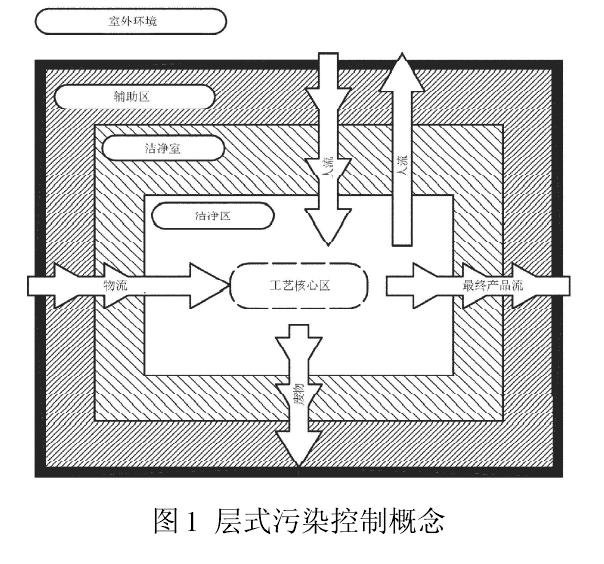
GB/T25915.4(2010)有(yǒu)着与新(xīn)版 GMP 同样的控制理(lǐ)念,都為(wèi)了有(yǒu)效控制药品生产环境的污染。图 1 是 GB/T25915.2010)的层式污染控制概念。工艺核心區(qū)(与环境发生交互作用(yòng)的工艺位置)所在的區(qū)域為(wèi)洁净區(qū),是洁净室中受控最严的部分(fēn),空气洁净度等级為(wèi)单向流100 级(即ISO 5 级,欧盟 A 级)。出于经济、技术和运行等方面的原因,要尽量减小(xiǎo)最高洁净度區(qū)域的范围。洁净區(qū)通常采用(yòng)密闭式(如 RABS),或由洁净度较低的外部區(qū)域-洁净室(1 万级,即 ISO7 级)包围。相邻區(qū)域间的人流、物(wù)流进入工艺核心區(qū)时,会增加传播污染的风险,因此应特别注意人流和物(wù)流的布局细节与管理(lǐ),这就是层式污染控制的基本概念。
与 GB/T25915.4(2010)不同的是新(xīn)版GMP将包围洁净區(qū)的外部區(qū)域(洁净室)的空气洁净度等级设定為(wèi)B级,所谓B级是静态控制时相同于A 级(单向流 100 级)的静态,动态控制时相同于 C 级(1 万级)的静态。GB/T 25915.4(2010)和新(xīn)版 GMP 都认為(wèi)无菌灌装線(xiàn)外部區(qū)域(洁净室)空气洁净度等级应低于工艺核心區(qū)(洁净區(qū)),至于低多(duō)少,是1 万级还是B 级?这是可(kě)探讨的學(xué)术问题,不应成為(wèi)判定标准高低的分(fēn)界線(xiàn)。國(guó)内外实践证明,背景设置為(wèi)1 万级或B 级,都能(néng)保护无菌药品的生产环境,我们应从分(fēn)析影响无菌灌装線(xiàn)的环境因素着手,寻找控制污染源的途径,才能(néng)最终确保无菌生产环境。
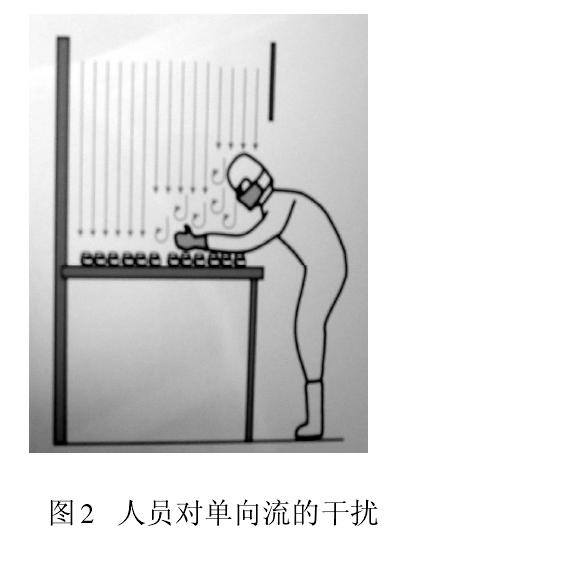
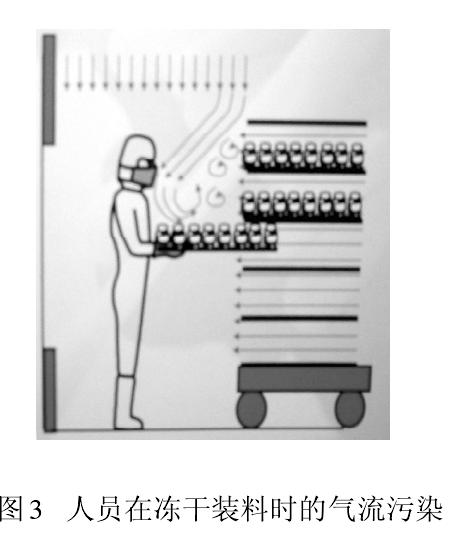
从人们开始接受并应用(yòng)洁净技术时就发现,人是洁净室最大的污染源。以微粒和微生物(wù)為(wèi)污染物(wù)控制对象的制药生产,大量测试数据和生产实践告诫我们,人员(生产、检验、管理(lǐ)、维修)因素无时无刻不在干扰药品生产。图2 显示人员在单向流下操作导致气流粒子污染。此刻,保护性的单向流变成污染物(wù)的载體(tǐ),直接影响被保护的产品。此时气流速度越大,则污染程度越大。图3 為(wèi)冻干剂装料时,单向流罩下垂直和水平气流相互冲突,
形成紊流,紊流气流会在较長(cháng)时间里产生悬浮污染。
不言而喻,人是无菌灌装过程中影响最大的环境因素。為(wèi)此,实现无菌灌装环境控制的技术要点是:(1)将人员从无菌环境隔离,从根本上排除人的干扰;(2)采取局部隔离无菌环境 (如R A B S 等);(3 )即便如此仍需限制人员对无菌产品的干预;(4)生产运行中不让人员进入无菌环境。不解决人对无菌环境干扰问题,大幅度提高无菌药品生产線(xiàn)外部區(qū)域空气洁净度等级,效果也不会明显。
必须强调,医药工程设计中空气净化措施只是控制洁净室微生物(wù)的手段之一,这些措施只能(néng)控制通过空气途径传播的微生物(wù)污染,对表面对象微生物(wù)的生物(wù)污染以及化學(xué)污染都无能(néng)為(wèi)力,药品生产使用(yòng)的设备、设施、容器具、物(wù)料、包装材料等沾附的微生物(wù)、化學(xué)品,以及生产、管理(lǐ)人员携带的微生物(wù),只有(yǒu)采用(yòng)清场、清洗、消毒、灭菌等措施,任何级别的净化空调都无法替代。
近年来國(guó)外对空气净化的滤菌效果提出质疑。数据表明 1 台
净技术老路,将会大大浪费洁净室建设费用(yòng)和运行成 本 。
國(guó)标 GB/T 25915.4(2010)的内容包括规划和设计、建造和启动、检测和验收、文(wén)件等方面要求,涵盖工程建设的全过程,对我國(guó)医药行业尤為(wèi)重要。药品生产实施 GMP 是一项系统工程,為(wèi)药品生产提供符合 GMP 要求的厂房设施是企业实施G M P 的先决条件。因此,医药工程项目必须按GMP 要求实施规范管理(lǐ),才能(néng)為(wèi)企业实施 GMP提供可(kě)靠保障。近年来國(guó)外针对医药工程项目的规范化管理(lǐ),一项新(xīn)的管理(lǐ)规范― ― GEP已在一些发达
國(guó)家悄然问世。GEP(医药工程质量管理(lǐ)规范)是Good Engineering Practice 的缩写,是与GMP配套的药品生产管理(lǐ)文(wén)件。它的宗旨是“将已确立的工程设计方案和标准应用(yòng)于项目的整个生命周期中,提供合适成本的解决方案”。我國(guó)至今尚无这方面的规范,工程项目大多(duō)由企业凭经验自行管理(lǐ),主要依据是各类相关的國(guó)家标准,缺乏系统性、协调性。
國(guó)标 GB/T 25915.4(2010)的发布,為(wèi)工程项目规划、设计、施工、试車(chē)、检测、验收等环节的规范化管理(lǐ)填补了空白,國(guó)标所附的大量资料性附录,如“设施的验收”、“设施的布局”、“建造和材料”、“洁净室的环境控制”、“待需方/ 用(yòng)户与供方/ 设计方商(shāng)定的补充技术要求”等,為(wèi)项目的工程设计、采購(gòu)供应、施工安装、试車(chē)验收、工程进度、技术质量、安全环保、资金运作、部门协调等提供了详尽的规范要求,制药企业应自觉以國(guó)标GB/T 25915為(wèi)准绳,并据此制订医药工程项目的GEP,确保工程项目符合 GMP 要求。
國(guó)标GB/T 25915系列标准反映了当今國(guó)际“洁净室及相关受控环境 ”的新(xīn)概念、新(xīn)思路和新(xīn)方法,是指导我國(guó)各行各业洁净技术的规范性文(wén)件,必将成為(wèi)我國(guó)洁净技术蓬勃发展的新(xīn)起点。
本文(wén)标签:洁净室
地址:山(shān)东省济南市天桥區(qū)新(xīn)徐居委会黄河建邦大桥西侧1-6号 電(diàn)话:0531-68824415 传真:0531-68824415
版权所有(yǒu):济南顺奇净化工程有(yǒu)限公司 技术支持:康美科(kē)技 备案号: html地图 登录